Genomic Data Analysis
The next-generation sequencing method has made it possible to produce large-volume genomic data as
the
technology advances and the analysis cost decreases rapidly in comparison with Moore’s law.
Accordingly, it is
vital to accurately detect variations related to a certain disease based on the
generated genomic data and to
determine the proper diagnosis, treatment method, and relevance to the efficacy.
Genome data flow diagram using NGS
-
Blood or FFPE
Sample Library
Library
Preparation and
CaptureSequencer NGS Data
NGS Data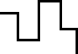
Upload -

-
Data analysisBioinformatics

Pipeline (Variant detection)Visualization
InterpretationReport
Variant detection
Difficulty in detecting variants in clinical settings
Genetic variants that can be detected through the analysis pipeline include single nucleotide variants (SNV), Insertion/Deletion (INDEL), copy number variants (CNV), structural variants (SV), etc.
Mutation detection does not end with simple detection, but it is important to detect a type of mutation with clinical significance. For example, in the case of CEBPA, a major blood cancer gene, a combination of bi-allelic mutations, in which mutations appear simultaneously at the N-terminal and C-terminal, should be applied for detection, rather than simple SNV/INDEL detection.
Accurate mutation detection requires the development and application of mutation detection algorithms using actual clinical sample data, and must be verified with numerous data.
In the case of NGS genomic data, there may be False Positive (FP) variants depending on the sequencing structure of genes and panels. Such variations cause challenges in the filtering process after detection.
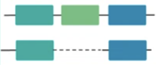
Insertion, deletion, duplication,
or inversion sequence variants
15-49 bp in size.
Examples:
CDKN2A:c.9_32dup
DHCR7:c.385_412+5del
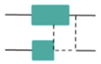
A CNV spanning a single exon,
part of one exon, or a single plus a
partial exon.
Examples:
DMD: Gain exon 22
FLCN: Deletion exon 14
associated
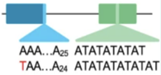
Variant in or near a homopolymer, STR, or
low-complexity region. Variant may or
may not be a simple length polymorphism.
Examples:
MSH2.c.942+3А>T
SCN1A:c.3724_3725dup
Lincoln, S.E., Hambuch, T., Zook, J.M. et al. One in seven pathogenic variants can be challenging to detect by NGS: an analysis of 450,000 patients with implications for clinical sensitivity and genetic test implementation. Genet Med 23, 1673–1680 (2021). https://doi.org/10.1038/s41436-021-01187-w
Enhancement of the variation detecting technology through the clinical analysis know-how
For major variations hardly detected and related to diseases such as CEBPA bi-allelic mutation, FLT3-ITD, MET exon14 skipping, 1p/19q co-deletion, etc., the detecting accuracy has been upgraded based on the know-how accumulated through analysis of actual clinical samples.
In addition, information on molecular signatures in various cancer types such as homologous recombination deficiency (HRD), microsatellite instability (MSI), tumor mutational burden (TMB), etc. is also provided, or related R&D projects are in progress.
By processing repeated FP variations, accurate variation results are made available to users.
The variation-detecting technology is improving based on the continued application and verification of the latest analysis technology.
Variant analysis
Now the focus is not merely on variant detection but on the accurate interpretation of detected variations.
Numerous variants have been detected based on the analysis of large-volume NGS data. Detected variants need to be accurately interpreted and documented.
To avoid confusion in reporting variants and exchanging information, all variants are accurately described according to the nomenclature of the Human Genome Variation Society (HGVS), an international standard nomenclature.
Accurate variant interpretation is implemented by integrating various types of databases such as disease-specific databases, variation distribution databases, etc.
Pathogenicity interpretation of mutations is provided by applying the pathogenicity determination guidelines for gene mutations announced by the American Society of Medical Genetics (ACMG), Society of Molecular Pathologists (AMP), Society of Clinical Oncology (ASCO), and Society of Pathologists (CAP).
Challenges in Variation Interpretation
There is little difficulty in the pathogenic interpretation of variants or genes that have already been extensively researched.
However, it is still challenging to interpret variants that have not been researched and therefore do not have much information available.
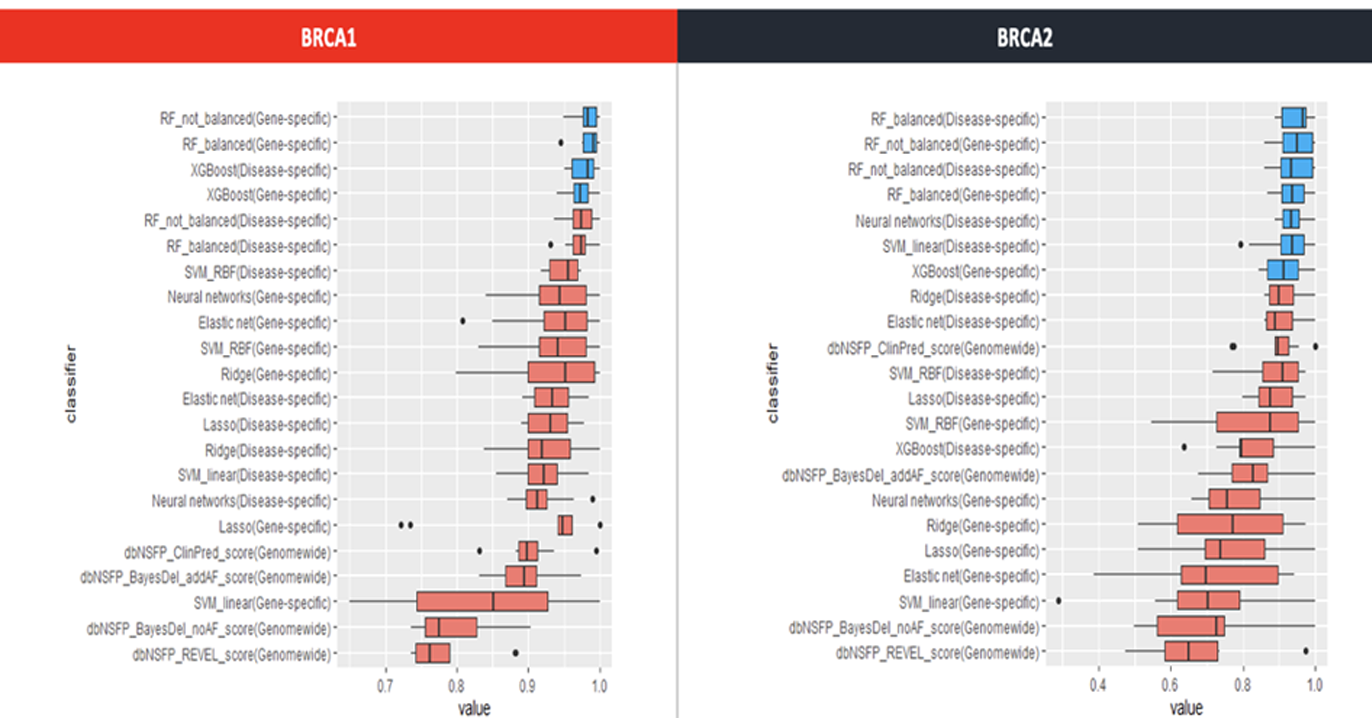
Machine learning to address such challenges in variation interpretation
As machine learning technology advances, this technology can be applied to variant interpretation as well.
Even for variants with little information, pathogenic prediction is possible by applying a self-developed machine learning model.