유전체 데이터 분석
차세대염기서열분석법은 기술의 발달과 더불어 분석 비용이 무어의 법칙보다 빠르게 감소하여 대용량 유전체 데이터 생산이 가능하게 되었으며,
이를 통해 생성한 유전체 데이터에서 질병과 연관된 변이를 정확히 찾고, 이 변이들이 진단, 치료 결정, 예후에 관련성이 있음을 찾아내는 분석과정이 중요합니다.
NGS를 이용한 유전체 데이터 흐름도
-
Blood or FFPE
Sample Library
Library
Preparation and
CaptureSequencer NGS Data
NGS Data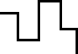
Upload -

-
데이터 분석변이 검출(Bioinformatics

Pipeline)변이 해석(Visualization
Interpretation)결과지(Report)
변이 검출
임상에서의 변이검출 어려움
분석 파이프라인 결과로 확인할 수 있는 변이는 Single nucleotide variants(SNV), Insertion/Deletions(INDEL),
Copy number variants(CNV), Structural variants(SV) 등이 있습니다.
변이 검출은 단순 검출에서 끝나는 것이 아니라, 임상적인 의의가 있는 변이 유형으로의 검출이 중요합니다.
예를 들어 혈액암 주요 유전자인 CEBPA 같은 경우 단순 SNV/INDEL 검출이 아닌, N-terminal, C-terminal 에서
동시에 변이가 나타나는 bi-allelic mutation 조합이 검출에 적용되야 합니다.
정확하게 변이를 검출하기 위해서는 임상 샘플 데이터를 활용하여 변이 검출 알고리즘을 개발해야 하며,
수 많은 임상 샘플 데이터로 분석하면서 지속적인 보완 작업이 진행되어야 합니다.
차세대 염기서열 데이터의 경우에는 패널 및 유전자의 서열 특성에 따라 False Positive(FP) variants 가 나타날 수 있기 때문에,
이러한 변이들을 제거하는 과정은 숙련된 기술이 필요합니다.
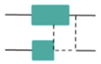
Insertion, deletion, duplication,
or inversion sequence variants
15-49 bp in size.
Examples:
CDKN2A:c.9_32dup
DHCR7:c.385_412+5del
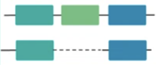
A CNV spanning a single exon,
part of one exon, or a single plus a
partial exon.
Examples:
DMD: Gain exon 22
FLCN: Deletion exon 14
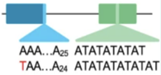
associated
Variant in or near a homopolymer, STR, or
low-complexity region. Variant may or
may not be a simple length polymorphism.
Examples:
MSH2.c.942+3А>T
SCN1A:c.3724_3725dup
Lincoln, S.E., Hambuch, T., Zook, J.M. et al. One in seven pathogenic variants can be challenging to detect by NGS: an analysis of 450,000 patients with implications for clinical sensitivity and genetic test implementation. Genet Med 23, 1673–1680 (2021). https://doi.org/10.1038/s41436-021-01187-w
임상 분석 노하우를 통한
변이 검출 기술 향상
질병과 연관된 중요 변이인 CEBPA bi-allelic mutation, FLT3-ITD, MET exon14 skipping, 1p/19q co-deletion 등
검출이 어려운 변이들에 대한 검출 정확도를 실제 임상 샘플을 분석하며 쌓인 노하우를 통해 높였습니다.
이 외 여러 암종에서 발생하는 분자 시그니처(Molecular signature)인 상동재조합결핍(HRD), 현미부수체 불안정성(MSI),
종양 돌연변이 부하(TMB)와 같은 결과를 제공 또는 연구개발 중 입니다.
반복 되는 FP 변이에 대한 처리를 적용하여 정확한 변이 결과를 사용자에게 제공하고 있습니다.
최신 분석 기술의 지속적인 적용 및 검증을 통해 변이 탐지 기술을 향상시키고 있습니다.
변이 해석
이제 중요한 것은 변이를 탐지하는 것이 아닌,
탐지된 변이들을 정확하게 해석하는 것입니다.
대용량의 유전체 데이터가 분석되면서 수많은 변이들이 탐지되었습니다. 탐지된 변이는 정확하게 해석되어야 하고, 기록되어야 합니다.
변이를 보고하고 정보를 교류하는 데 있어서 혼란을 주지 않기 위해, 모든 변이는 국제 표준 명명법인 Human Genome Variation Society (HGVS) 의 명명법에 따라 정확하게 기술되어야 합니다.
질병 특이적 데이터베이스와 변이 분포 데이터베이스 등 여러 종류의 데이터베이스들을 종합하여 정확한 변이 해석을 진행해야 합니다.
미국의 의학유전학회(ACMG), 분자병리학회(AMP), 임상종양학회(ASCO), 병리학회(CAP) 등에서 발표한 유전자 변이에 대한 해석 지침에 따라 변이를 해석해야 합니다.
변이 해석에 있어서 어려운 점
연구가 많이 진행된 유전자 또는 변이에 대한 임상 해석은 문제가 없습니다.
하지만 연구가 진행되지 않거나 정보가 많지 않은 변이에 대해서는 해석하는 데 여전히 어려움이 있습니다.
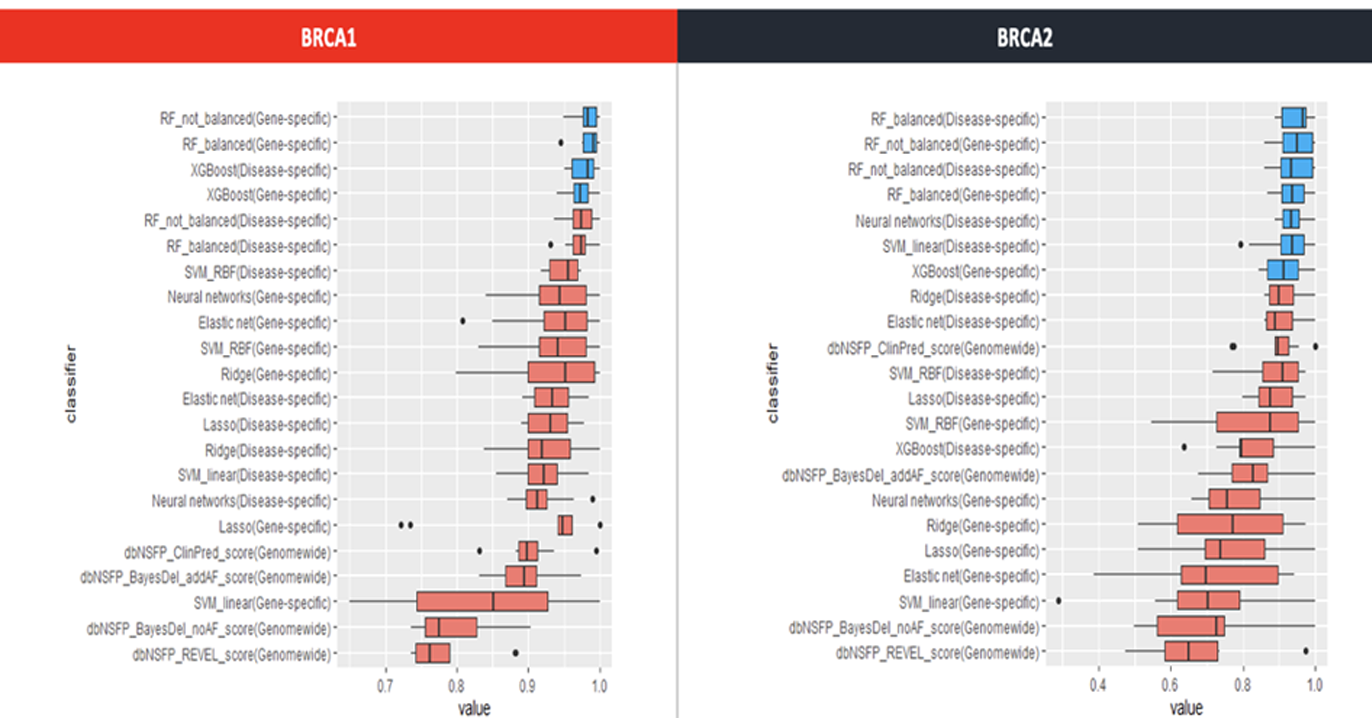
변이 해석의 어려움을 머신러닝을 통해 해결
머신러닝 기술이 발전을 이루면서, 변이 해석에도 해당 기술을 적용하여 해석을 진행할 수 있습니다.
정보가 많지 않은 변이일지라도, 자체 개발한 머신러닝 모델을 이용하여 병원성을 예측할 수 있게 되었습니다.